Bloom filters for bioinformatics
- Abhishek Tiwari
- Research
- 10.59350/1082w-a3b44
- Crossref
- June 21, 2010
Table of Contents
The Bloom filter was originally developed by Burton H. Bloom back in the seventies and for long time it was there without any major application. Google is credited for making Bloom filter popular again. Only after the Google used Bloom filters for their BigTable database system, the idea started grabbing the attention of larger and diverse audience.
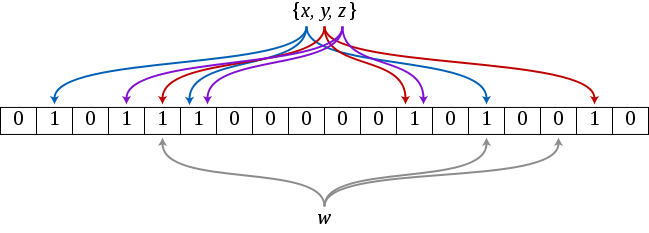
Bloom filter is an extremely space-efficient probabilistic data structure which enables the quick and easy membership tests. On positive side, in order to test whether or not an element is a member of a set, Bloom filters need less memory than any other data structure such as hash tables, simple arrays or linked lists. On downside, the risk of false positives is higher.
Any task that require checking two sets of data against each other can be performed in an efficient way using Bloom filter. For instance, one can use Bloom filter to do spell-checking against a dictionary of correct words.
Recently Bioinformatics community started using Bloom filters for large scale gene sequence analysis. Comparing sequences to test similarity is a common task in bioinformatics. For instance,
one might want to know where a certain gene is located in the chromosome, or which sequence fragments are similar enough to originate from the same gene. To speed up searches, it is common to index sequences in questions as overlapping, substrings (k-tuples, q-grams). This index seems like an obvious target for Bloom filters — large data, time critical, some false positives anyway — but for some reason, there is almost no such applications that use them. Until now.
In a recent paper in journal Bioinformatics, Stranneheim et. al describe a novel Bloom filters based algorithm, FACS (Fast and Accurate Classification of Sequences) for accurate and rapid classification of DNA sequences as belonging or not belonging to a reference DNA sequence. In this case reference DNA sequence can be as large as the whole genome. This kind of rapid classification method can be a boon for metagenomic studies where one need to classify sequences as ’novel’, or belonging to a well known genome.
A comparative study using metagenomic data sets suggest that FACS method is at least 21 times faster compared to algorithms such as BLAT and SSAHA2 with nearly same accuracy. The FACS algorithm is implemented as PERL module and it can be downloaded from CPAN.
Similarly Malde and O’Sullivan have developed some interesting bloom filter based sequence analysis applications in Haskell. In their analysis they matched randomly selected ESTs against the E. coli genome which is relatively small compared to human genome.
In terms of memory consumption their Bloom filter application was using a mere 20MB, of which the Bloom filter itself needed only 2MB compared to the set based implementations those allocated 160-190MB of memory for a small test case.
As such its very easy to implement your own Bloom filter, but you can enjoy existing Bloom filter implementations in various languages.
- Bloom filter using C++
- Bloom filter using C
- Bloom filter using Ruby (gem install bloomfilter, BloominSimple, sBloomFilter, Ruby Counting Bloom Filter )
- Bloom filter using Haskell
- Bloom filter using Java

